Chondrosarcoma
| Chondrosarcoma | |
|---|---|
| Diagnosis in short | |
|
Template:Px Chondrosarcoma. H&E stain. | |
|
| |
| LM | "abnormal cartilage": +/-high grade changes - nuclear atypia (nuclear clearing, nucleoli, hyperchromasia), low/intermediate grade changes - bi-nucleation, hypochromatic enlarged nuclei, infiltration of lamellar bone ("invasion"), increased cellularity, irregular spacing of chondrocytes |
| Subtypes | chondrosarcoma not otherwise specified (NOS), juxtacortical chondrosarcoma, myxoid chondrosarcoma, mesenchymal chondrosarcoma, clear cell chondrosarcoma, dedifferentiated chondrosarcoma |
| LM DDx | chondroblastic osteosarcoma, enchondroma (esp. for low-grade chondrosarcoma), chordoma, others |
| Molecular | t(9;22) for extraskeletal myxoid chondrosarcoma |
| Gross | cartilaginous appearance |
| Site | hip, shoulder, soft tissue, others |
|
| |
| Syndromes | Olier disease, Maffucci syndrome |
|
| |
| Clinical history | adults |
| Signs | mass lesion |
| Prevalence | uncommon |
| Prognosis | good ~75% five year survival |
| Clin. DDx | enchondroma, bone tumours, soft tissue lesions |
| Treatment | excision |
Chondrosarcoma is a malignant tumour of cartilage. It is in the chondro-osseous grouping of tumours and can be lumped into the much large category of the soft tissue lesions.
General
- Usually a good prognosis - 75% five year survival in one large data set.[1]
- Subtypes vary substantially - chondrosarcoma NOS and myxoid chondrosarcoma have a five year survival of ~70%, but mesenchymal chondrosarcoma only ~50%, and dedifferentiated chondrosarcoma an abysmal ~0%![2]
- Grade and stage are independent predictors of survival.[2]
Clinical/epidemiologic features:[3]
- Usually arise in a (benign) abnormality of cartilage (e.g. osteochondroma, enchondroma).
- May be associated with a syndrome:
- Olier disease (multiple enchondromatosis).
- Maffucci syndrome (multiple enchondromas and hemangiomas).
Subtypes
Several subtypes and their relative prevalence:[2]
- Chondrosarcoma not otherwise specified (NOS) ~83% of cases.
- Juxtacortical chondrosarcoma <1% of cases.
- Myxoid chondrosarcoma ~10% of cases.
- Mesenchymal chondrosarcoma ~4% of cases.
- Clear cell chondrosarcoma <1% of cases
- Dedifferentiated chondrosarcoma ~1% of cases.
Gross
- Appendicular skeleton ~45% of cases.[2]
- Classically hip.
- Axial skeleton ~30% of cases.
- Soft tissue ~10% of cases.
Note:
- Peripheral chondrosarcoma are very rare.[4]
- Chondrosarcoma is the most common primary malignant chest wall lesion.[5]
- The classical location is anterior (costochondral arches or sternum), where it is more common than chondroma.
Microscopic
- "Abnormal cartilage":
- +/-Nuclear atypia - high grade lesions.
- High grade lesions:
- Nuclear clearing.
- Nucleoli.
- Hyperchromasia.
- Low/intermediate grade lesions:
- Bi-nucleation.
- Hypochromatic enlarged nuclei.
- Infiltration of lamellar bone ("invasion") - not common - diagnostic.
- High grade lesions:
- Increased cellularity.
- More cellular than cartilage... but relatively paucicellular compared to other sarcomas.
- Irregular spacing of chondrocytes.
- +/-Nuclear atypia - high grade lesions.
Notes:
- Low grade chondrosarcoma are not cytologically malignant; the diagnosis rests mostly on radiologic findings.
- The exception is infiltration of lamellar bone -- this is diagnostic of chondrosarcoma.[8]
DDx:
- Chordoma.
- Enchondroma.
- Synovial chondromatosis.
- Osteosarcoma - esp. chondroblastic osteosarcoma - has osteoid, may be focal.
Images
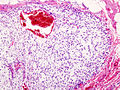
Chondrosarcoma - low mag. (WC)
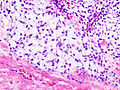
Chondrosarcoma - high mag. (WC)
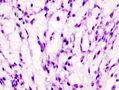
Chondrosarcoma - high mag. (WC)
Grade 2 chondrosarcoma, low power showing bone permeation (SKB)
Chondrocytic hyperchromasia and atypicality in this photo of a Grade 2 chondrosarcoma. (SKB)
Lobule of neoplastic Grade 2 cartilage with some bone permeation and calcification at the top of the photo.(SKB)
Lobule of neoplastic Grade 2 cartilage with atypia and bone permeation. (SKB)
- Bone Chondrosarcoma Grade3 MP PA.JPG
Grade 3 - very cellular neoplastic cartilage with high grade nuclear atypia. (SKB)
.jpg)
.jpg)
.jpg)
www:
%20(Large).jpg){kind=link}
Variants
Mesenchymal chondrosarcoma
Myxoid chondrosarcoma
Microscopic: Features:
DDx:
- Chondroid syringoma - These are dermal based, circumscribed and much smaller.
- Parachordoma.[10]
- Chordoma. (???)
- Myxoid liposarcoma.
- Metastatic myxoid carcinoma.
Images:
- Bone Chondrosarcoma Myxoid MP2 PA.JPG
Anastomizing chords of small neoplastic cells surround mucin pools.(SKB)
- Bone Chondrosarcoma Myxoid MP PA.JPG
Chords of neoplastic cells surround mucin pools. (SKB)
![[1]](http://pathologyoutlines.com/wick/softtissue/chondrosarcomaextraskeletalmyxoidtypemicro1.jpg){kind=link}
![[2]](http://pathologyoutlines.com/wick/softtissue/chondrosarcomaextraskeletalmyxoidtypemicro2.jpg){kind=link}
Clear cell chondrosarcoma
- Rare variant of chondrosarcoma (1.6%–5.4% of all chondrosarcomas)
- Usually a low-grade malignant tumour[11]
- Younger age than conventional chondrosarcoma
- Teens to 40s; more common in males
- Epiphyses of long tubular bones; proximal femur or humerus
- Rarely head and neck [12]
- Malignant counterpart of chondroblastoma?
Microscopic findings
- Lobules of uniform to polymorphic densely-packed large cells
- Well defined pushing borders
- Clear to intensively acidophilic granular cytoplasm cytoplasm with vacuoles
- Central nuclei with occasional prominent nucleoli
- Low mitotic rate
- Clear cell areas lack production of hyaline chondroid matrix
- Areas with osteoclast-type giant cells mixed with small trabeculae of reactive bone
- May contain conventional low-grade chondrosarcoma
- May have secondary aneurysmal bone cyst changes
- Bone Chondrosarcoma ClearCell HP PA.jpg
High grade round cells with cytoplasmic clearing. (SKB)
- Bone Chondrosarcoma ClearCell MP3 PA.jpg
High grade round cells with cytoplasmic clearing. (SKB)
- Bone Chondrosarcoma ClearCell MP4 PA.jpg
Clear cells, giant cells and bone spicules. (SKB)
- Bone Chondrosarcoma ClearCell MP2 PA.jpg
Somewhat hemangiopericytomatous vascular pattern with giant cells. (SKB)
- Bone Chondrosarcoma ClearCell MP4 PA.jpg
Clear cells, giant cells and bone spicules. (SKB)
- Bone Chondrosarcoma ClearCell MP PA.jpg
The lesion also had areas of more conventional chondrosarcoma. (SKB)
- Pathology outlines [3]
- Pathology outlines [4]
- Pathology outlines [5]
- Tumor library [6]
- Tumor library [7]
![[3]](http://pathologyoutlines.com/wick/chondrosarcoma%20clear%20cell%20type%20micro7.jpg){kind=link}
![[4]](http://pathologyoutlines.com/wick/chondrosarcoma%20clear%20cell%20type%20micro8.jpg){kind=link}
![[5]](http://pathologyoutlines.com/wick/chondrosarcoma%20clear%20cell%20type%20micro2.jpg){kind=link}
![[6]](http://www.tumorlibrary.com/case/images/1521.jpg){kind=link}
![[7]](http://www.tumorlibrary.com/case/images/1522.jpg){kind=link}
DDX:
- Chondroblastoma
- Giant cell tumour of bone
- Osteoblastic tumours
- Metastatic clear cell renal cell carcinoma
Outside sources: E-immunohistochemistry[8]
Extraskeletal myxoid chondrosarcoma
- Originally thought to be a variant of myxoid chondrosarcoma of bone; however, may not be a chondrosarcoma at all.[13]
- Characteristic chromosomal translocation: t(9;22) CHN-EWS.
DDx:
- Chordoma.[13]
- S-100 +ve (strong).
- EMA +ve.
- Myxoid liposarcoma.
Image:
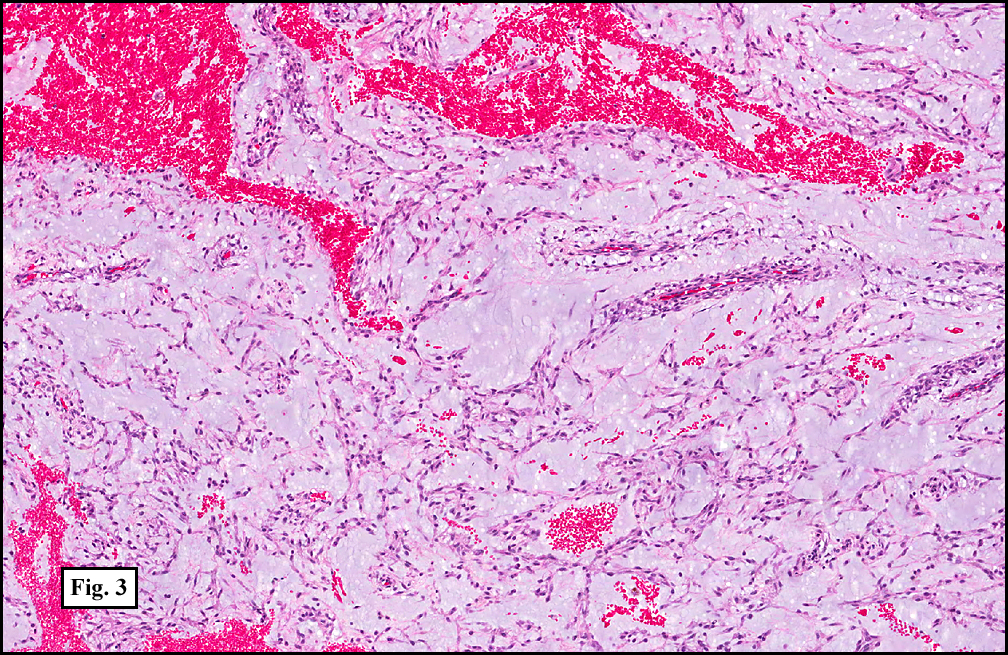{kind=link}
- SoftTissue ExtraskeletalMyxoidChondrosarcoma MP CTR.jpg
Strands of atypical cells suspended in mucin. (SKB)
- SoftTissue ExtraskeletalMyxoidChondrosarcoma HP CTR.jpg
Strands of atypical cells suspended in mucin. (SKB)
Dedifferentiated chondrosarcoma
Clinical:
- Abysmal to poor prognosis.
Features:[16]
- Poorly differentiated (mesenchymal) malignancy.
- Well-differentiated cartilaginous component.
DDx:
- Undifferentiated pleomorphic sarcoma - no cartilaginous component.
- Other dedifferentiated tumours, e.g. dedifferentiated liposarcoma, with a minimal differentiated component.
Images:
- Bone Chondrosarcoma Dedifferentiated PA copy.jpg
A lobule of cartilagenous chondrosarcoma on the left; high grade sarcoma on the right. (SKB)
- Bone Chondrosarcoma Dedifferentiated HP PA.jpg
High grade sarcoma on the left and upper; malignant cartilage right and lower. (SKB)
Grading
Features:[17]
- Grade I: mild-to-moderate increase of cellularity +/- binucleated cells.
- Grade II: between Grade I and Grade III.
- Grade III: nuclear pleomorphism, mitoses common.
Grade 1 - Somewhat cellular cartilage with binucleation.(SKB)
Grade 2 - Very cellular cartilage with obvious hyperchromasia and nuclear atypia. (SKB)
- Bone Chondrosarcoma Grade3 HP PA.JPG
Grade 3 - Even more cellular neoplastic cartilage with high grade nuclear atypia. (SKB)
IHC
- S-100 positive
- Keratin negative
- Collagen II positive
See also
References
- ↑ Damron, TA.; Ward, WG.; Stewart, A. (Jun 2007). "Osteosarcoma, chondrosarcoma, and Ewing's sarcoma: National Cancer Data Base Report.". Clin Orthop Relat Res 459: 40-7. doi:10.1097/BLO.0b013e318059b8c9. PMID 17414166.
- ↑ 2.0 2.1 2.2 2.3 Giuffrida, AY.; Burgueno, JE.; Koniaris, LG.; Gutierrez, JC.; Duncan, R.; Scully, SP. (May 2009). "Chondrosarcoma in the United States (1973 to 2003): an analysis of 2890 cases from the SEER database.". J Bone Joint Surg Am 91 (5): 1063-72. doi:10.2106/JBJS.H.00416. PMID 19411454.
- ↑ Skubitz KM, D'Adamo DR (November 2007). "Sarcoma". Mayo Clin. Proc. 82 (11): 1409–32. PMID 17976362. http://www.mayoclinicproceedings.com/content/82/11/1409.long.
- ↑ Henderson, ER.; Pala, E.; Angelini, A.; Rimondi, E.; Ruggieri, P. (2013). "Dedifferentiated peripheral chondrosarcoma: a review of radiologic characteristics.". Sarcoma 2013: 505321. doi:10.1155/2013/505321. PMID 23589702.
- ↑ Somers, J.; Faber, LP. (Jul 1999). "Chondroma and chondrosarcoma.". Semin Thorac Cardiovasc Surg 11 (3): 270-7. PMID 10451259.
- ↑ IAV. 26 February 2009.
- ↑ Klatt, Edward C. (2006). Robbins and Cotran Atlas of Pathology (1st ed.). Saunders. pp. 417. ISBN 978-1416002741.
- ↑ Dickson, B. 28 April 2011.
- ↑ URL: http://www.path.utah.edu/casepath/ms%20cases/MSCase6/MSCase6Part3.htm. Accessed on: 29 December 2013.
- ↑ Fisher C (May 2000). "Parachordoma exists--but what is it?". Adv Anat Pathol 7 (3): 141–8. PMID 10809219.
- ↑ Corradi, D.; Bacchini, P.; Campanini, N.; Bertoni, F. (Nov 2006). "Aggressive clear cell chondrosarcomas: do distinctive characteristics exist?: a report of 4 cases.". Arch Pathol Lab Med 130 (11): 1673-9. doi:10.1043/1543-2165(2006)130[1673:ACCCDD]2.0.CO;2. PMID 17076530.
- ↑ Mokhtari, S.; Mirafsharieh, A. (2012). "Clear cell chondrosarcoma of the head and neck.". Head Neck Oncol 4: 13. doi:10.1186/1758-3284-4-13. PMID 22520362.
- ↑ 13.0 13.1 Aigner, T.; Oliveira, AM.; Nascimento, AG. (Feb 2004). "Extraskeletal myxoid chondrosarcomas do not show a chondrocytic phenotype.". Mod Pathol 17 (2): 214-21. doi:10.1038/modpathol.3800036. PMID 14657948.
- ↑ URL: http://www.cttr.org/cms/?p=736. Accessed on: 1 May 2011.
- ↑ Mitchell, AD.; Ayoub, K.; Mangham, DC.; Grimer, RJ.; Carter, SR.; Tillman, RM. (Jan 2000). "Experience in the treatment of dedifferentiated chondrosarcoma.". J Bone Joint Surg Br 82 (1): 55-61. PMID 10697315.
- ↑ 16.0 16.1 Sopta, J.; Dordević, A.; Tulić, G.; Mijucić, V. (Feb 2008). "Dedifferentiated chondrosarcoma: our clinico-pathological experience and dilemmas in 25 cases.". J Cancer Res Clin Oncol 134 (2): 147-52. doi:10.1007/s00432-007-0262-5. PMID 17653766.
- ↑ Humphrey, Peter A; Dehner, Louis P; Pfeifer, John D (2008). The Washington Manual of Surgical Pathology (1st ed.). Lippincott Williams & Wilkins. pp. 643. ISBN 978-0781765275.