Difference between revisions of "Neurodegenerative diseases"
Jensflorian (talk | contribs) (→Atypical FTLD‐U: update) |
Jensflorian (talk | contribs) (→Overview: update) |
||
| Line 13: | Line 13: | ||
{{familytree | | | | | | | A01 | | | | | | | | A01=Neurodegenerative<br>disorders}} | {{familytree | | | | | | | A01 | | | | | | | | A01=Neurodegenerative<br>disorders}} | ||
{{familytree | |,|-|-|-|v|-|^|-|v|-|-|-|v|-|-|-|.| | |}} | {{familytree | |,|-|-|-|v|-|^|-|v|-|-|-|v|-|-|-|.| | |}} | ||
{{familytree | B01 | | B02 | | B03 | | B04 | | B05 || B01=Amyloidoses|B02=Tauopathies|B03=α-synucleinopathies|B04=TDP-43|B05=FUS}} | {{familytree | B01 | | B02 | | B03 | | B04 | | B05 || B01=Amyloidoses|B02=Tauopathies|B03=α-synucleinopathies|B04=TDP-43|B05=FUS/EWS/TAF15}} | ||
{{familytree/end}} | {{familytree/end}} | ||
| Line 36: | Line 36: | ||
*Frontotemporal lobar degeneration with TDP-43 (FTLD-TDP). | *Frontotemporal lobar degeneration with TDP-43 (FTLD-TDP). | ||
FET proteinopathies: | |||
*Basophilic inclusion body disease (BIBD). | *Basophilic inclusion body disease (BIBD). | ||
*Neuronal intermediate filament inclusion disease (NIFID). | *Neuronal intermediate filament inclusion disease (NIFID). | ||
* | *Atypical frontotemporal lobar degeneration with ubiquitin-positive inclusions (atypical FTLD-U). | ||
Prionopathies: | Prionopathies: | ||
Revision as of 14:03, 5 April 2019
Neurodegenerative diseases is a big part of neuropathology. It includes some discussion of dementia.
Overview
- Neurodegenerative disease = essentially progressive and selective neuron loss.
- Clinically, they are not unique, e.g. dementia can be caused by several diseases (with different molecular etiologies).
- Each syndrome (e.g. dementia, parkinsonism, ataxia) has a most common etiology and a DDx.
- They are defined by molecular pathology.[1]
- The diseases are due to the accumulation of abnormal protein.
- The amino acid sequence of the protein may be completely normal. The problem may just be folding/protein conformation.
- The diseases are due to the accumulation of abnormal protein.
Molecular schema of neurodegenerative disorders:[1]
| Neurodegenerative disorders | |||||||||||||||||||||||||||||||||||||||||
| Amyloidoses | Tauopathies | α-synucleinopathies | TDP-43 | FUS/EWS/TAF15 | |||||||||||||||||||||||||||||||||||||
Common diseases
- Alzheimer disease (Abeta).
'Pure' tauopathies:
- Progressive supranuclear palsy.
- Pick's disease.
- Corticobasal degeneration.
- FTDP-17.
- Dementia pugilistica.
Synucleinopathies:[2]
- Parkinson disease.
- Dementia with Lewy bodies.
- Multiple system atrophy.
TDP-43 proteinopathies:
- Amyotrophic lateral sclerosis.
- Frontotemporal lobar degeneration with TDP-43 (FTLD-TDP).
FET proteinopathies:
- Basophilic inclusion body disease (BIBD).
- Neuronal intermediate filament inclusion disease (NIFID).
- Atypical frontotemporal lobar degeneration with ubiquitin-positive inclusions (atypical FTLD-U).
Prionopathies:
- Creutzfeldt-Jakob disease (PrP).
Table
Disease/pathology/clinical correlation based on Dickson:[1]
| Disease | Mutated protein | Distribution | Clinical | Histology | Image |
|---|---|---|---|---|---|
| Alzheimer disease | Abeta (mutated APP) | corticolimbic, usu. spares occipital |
dementia | plaques, neurofibrillary tangles | [1] |
| Creutzfeldt-Jakob disease | PrPres (mutated PrP) | cortical & basal ganglia | dementia (rapid progression), movement disorder |
cytoplasmic vacuolization, PrP+ve plaques, Kuru plaques (MV2 variant) | [2] |
| Parkinson disease | alpha-synuclein | brainstem | parkinsonism | Lewy bodies in substantia nigra and locus coeruleus | [3] [4] |
| Dementia with Lewy bodies |
alpha-synuclein | corticolimbic, brainstem | dementia + parkinsonism | Lewy bodies brainstem and cortical, tangles | [5] [6] |
| Multiple system atrophy | alpha-synuclein | basal ganglia, brainstem, cerebellum | parkinsonism, ataxia | Papp-Lantos inclusions (cytoplasmic deposits in oligodendrocytes)[3] | [7] |
| Amyotrophic lateral sclerosis (ALS) |
TDP-43 | motor neurons | spasticity, weakness | motor neuron loss, TDP-43+ve, TAF15-ve, EWS-ve inclusions in motor neurons | [8] |
| Frontotemporal lobar degeneration with TDP-43 (FTLD-TDP) |
TDP-43 | cortex, basal ganglia | dementia, focal cortical syndromes | histology depends on (type 1-4), ubiquitin and TDP-43+ve, tau and FUS-ve | [9] |
| Frontotemporal lobar degeneration with FUS (FTLD-FUS) |
FUS | cortex, medulla, hippocampus, and motor cells of the spinal cord | dementia, cases classified as FTLD-U, NIFID and BIBD | FUS+ve, TAF15+ve, EWS+ve cytoplasmic & intranuclear inclusions, neuritic threads | [10] |
| Progressive supranuclear palsy (FTLD-tau) | tau 4R | basal ganglia, brainstem | atypical parkinsonism with early gait instability, falls, and supranuclear gaze palsy | tau-positive globose neurofibrillary tangles in neurons, tufted astrocytes, coiled bodies in oligodendrocytes |
[11] |
| Pick disease (FTLD-tau) | tau 3R | corticolimbic | dementia + focal cortical syndrome |
Intraneuronal argyrophilic inclusions (Pick body) | [12] |
| Corticobasal degeneration (CBD) (FTLD-tau) | tau 4R | cortical, basal ganglia | dementia + movement disorder (Parkinson-plus syndrome) | ballooned neurons, astrocytic plaques, pretangles in basal nucleus | [13] |
| Argryophilic grain disease (AGD) (FTLD-tau) | tau 4R | medial temporal lobe, limbic structures | late-onset amnestic syndrome | Argyrophilic grains (also found unspecific in elederly) | [14] |
Immunohistochemistry
Alpha-synuclein
Look for:
- Lewy bodies (seen in Parkinson's d., Dementia with Lewy bodies) = round cytoplasmic eosinophilic body +/- pale halo.
- Glial cytoplasmatic inclusions (Papp-Lantos bodies) seen in MSA.
Tau
TDP-43
- May accumulate due to a progranulin mutation.
Microscopic
- TDP-43 - normally in the nucleus.
- Pathologic: Micrograph (label B) - neurites, skein-like formations (ama-assn.org)[6]
- Fibrillar or skein-like formations = cytoplasmic staining.
- "Skein" = yarn or thread wound on a reel or flock of birds in flight.[7]
- Neurites = "squiggly appearance"; "worm-like appearance".
- Fibrillar or skein-like formations = cytoplasmic staining.
- Pathologic: Micrograph (label B) - neurites, skein-like formations (ama-assn.org)[6]
Ubiquitin
- Marks proteins for recycling.
Microscopic
- p62; poli-ubiquitin-binding protein p62.[4]
Look for:
- Lewy bodies. (???)
Clinical perspective
Dementia general (mostly useless) DDx
- Alzheimer's dementia - most common.
- Vascular.
- Multi-infarct dementia.
- Parkinson's associated dementia.
- Lewy body dementia.
- Alcohol-related dementia.
- Fronto-temporal dementia (Pick disease).
- Multisystem atrophy.
Mnemonic
Dementia mnemonic VITAMIN D VEST:[8]
- Vitamin deficiency (B12, folate, thiamine).
- Infection (HIV).
- Trauma.
- Anoxia.
- Metabolic (Diabetes).
- Intracranial tumour.
- Normal pressure hydrocephalus.
- Degenerative (Alzheimer's, Huntington's, CJD).
- Vascular.
- Endocrine.
- Space occupying lesion (chronic subdural hematoma).
- Toxins (alcohol).
Functional anatomy of dementia
- Hippocampus (essential for forming new memories).
- Frontal lobe (essential for retrieval of memories).
Parkinsonism causes
- Parkinson's disease [9]
- Dementia with Lewy bodies.
- Multiple system atrophy (MSA).[10]
- Progressive supranuclear palsy (PSP).[11]
- Drug induced (valproic acid, MPTP).[12][13]
- Vascular. [14]
- Postencephalitic. [15]
- Tramuatic (Dementia pugilistica).[16]
Amyloidoses
Alzheimer disease
General
- Onset: episodic memory loss.
- Diagnosis is clinical & pathologic.
- Pathologic finding alone are not diagnostic.
- Onset, rate of progression and the development of pathology are highly variable.
- Defined by:
- Pathological accumulation of amyloid β (Aβ) into extracellular plaques.
- Abnormally phosphorylated tau that accumulates intraneuronally forming neurofibrillary tangles (NFTs).
- Clinicopathological correlation better for NFT than for Aβ.[17]
- Seen in conjunction with vascular amyloid deposition; see cerebral amyloid angiopathy.
- Evidence of possible iatrogenic transmission by cadaver-sourced growth hormone batches.[18][19]
Genetics
Genes associated with Alzheimer disease:[20]
- Amyloid precursor protein (APP).
- On chromosome 21 - may explain why Trisomy 21 (Down syndrome) increases the risk of Alzheimer disease.[21]
- Presenilin 1 (PSEN1).[22]
- Presenilin 2 (PSEN2).[23]
- Apolipoprotein E (APOE)[24] - specifically the epsilon-4 allele.
Gross
Features:
- Temporal atrophy, esp. hippocampus.
- Dilation of:
- Lateral ventricles.
- Third ventricle.
Gross/microscopic - disease spread by NF tangles (staging):[25]
- Alzheimer "spreads" in a reproducible pattern:
- Stage I-II: entorhinal cortex.
- Stage III-IV: inferior aspect of brain.
- Stage V-VI: limbic system.
Minimal sampling:
- Frontal, parietal & temporal lobe
- Hippocampus and entorhinal cortex
Additional sampling:
- Basal ganglia
- Cerebellum
- Midbrain (including substantia nigra)
- Occipital cortex
Images
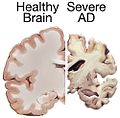
Alzheimer's brain. (WC/NIH)

Alzheimer's brain macroscopy (top) vs control (bottom). (WC/Hersenbank)

Alois Alzheimer provided a first description of the pathology. (NLM)



Microscopic
Features:
- Neurofibrillary tangles.
- Consists of tau.
- Location: hippocampus, cerebral cortex, hypothalamus.
- Dementia severity correlates better with NF tangles number than senile plaque number.[26]
- Six-tiered scoring method to assess tangle load [27]
- Images: tangles - schematic (pakmed.net)[28], tangle (washington.edu).[29]
- Senile plaques (AKA neuritic plaques).
- Consists of two components:
- Centre - radiates.
- Consists of Abeta amyloid
- Neurites - swollen axons.
- Centre - radiates.
- Considered to be more specific for Alzheimer's than NF tangles.
- How to remember: senile plaques = specific.
- There is a CERAD staging system for senile plaque load: 0 (none), I (mild), II (moderate), III (severe).[30]
- Images: senile plaques (utah.edu)[31] senile plaques - beta-APP - high mag. (WC).
- Consists of two components:
- Neuron loss.
- +/-Cerebral amyloid angiopathy.
Images
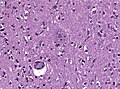
Neuritic plaques with amyloid core in HE. (WC/jensflorian)
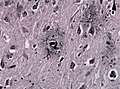
Neuritic plaques in Gallays silver impregnation. (WC/jensflorian)
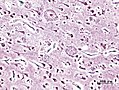
Neuritic plaques with amyloid core in Bodian stain. (WC/KGH)

Plaques with Abeta IHC. (WC/Nephron)
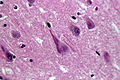
Neurofibrillary tangles in HE. (WC/Patho)
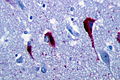
Tau IHC highlighting tangles. (WC/Patho)
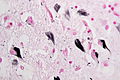
Tangles in Gallays silver impregnation.(WC/Patho)
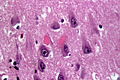
HE stain with ghost tangles and Hirano bodies in AD. (WC/Patho)
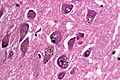
HE stain with granulovaculolar degeneration in AD. (WC/Patho)


_presenile_onset.jpg)






Classification
NIA/AA Guidelines: "ABC" scoring method [32]
- (A) assessment of amyloid b deposits
- (B) staging of neurofibrillary tangles
- (C) scoring of neuritic plaques
| (A) abeta plaques (Thal phase)[33] | (B) Neurofibrillary tangles (Braak stage) [34] | (C) neuritic plaques (CERAD) [35] |
|---|---|---|
| (A0) 0 | (B0) 0 | (C0) none |
| (A1) 1 (temporal),2 (+frontal, +CA1) | (B1) I,II (transentorhinal) | (C1) sparse (1–5 neuritic plaques/1 mm2) |
| (A2) 3 (+diencephalon, +striatum) | (B2) III,IV (limbic) | (C2) moderate(6–19 neuritic plaques/1 mm2) |
| (A3) 4 (+brainstem),5 (+cerebellum, +pons) | (B3) V,VI (neocortical) | (C3) frequent(>20 neuritic plaques/1 mm2) |
The ABC score is a good indicator for the likelihood of dementia.
Example: Cerebellar abeta deposits (A3) + tangles in entorhinal cortex and few temporal (B2), + 15 neuritic plaques per 1 mm2 (C2) -> (A3, B3, C2): intermediate AD level change.
Notes:
- Abeta amyloid:
- Derived from amyloid precursor protein (APP).
- APP:
- Rapid axonal transport - useful as a marker of axonal injury.
- Function currently not known.
- APP:
- Derived from amyloid precursor protein (APP).
- Tau:
- Important in microtubule assembly.
Prion diseases
General
Etiology:[36]
- Misfolded cell-surface protein called PrPSC.
- This is derived from the protein PrPC encoded by the PRNP gene.
Includes:
- Creutzfeldt-Jakob disease (CJD).
- Sporadic fatal insomnia (sFI).[36]
- Fatal familial insomnia (FFI).[37][38]
- Gestmann-Straussler-Scheinker syndrome (GSS) - due to PRNP gene mutations.[39]
IHC
PrPC:[37]
- Congo red +ve.
- PAS +ve.
Creutzfeldt-Jakob disease
- Commonly abbreviated as CJD.
General
- Rare.
- Incurable disease.
Usually diagnosed clinically:
- Characteristic findings:
- Very rapid decline (3-4 months).
- Characteristic (cortex findings on) neuroradiology.
Variant Creutzfeldt-Jakob disease
- Abbreviated vCJD.
General
- Associated with bovine spongiform encephalopathy (AKA mad cow disease).
- Should sample: spleen, lymph nodes, tonsils.[40]
Microscopic
Features:
- Spongy appearance (cytoplasmic vacuolization[41]).
Note:
- Spongiform changes may be seen in ALS, Alzheimer's disease and Lewy body disease (e.g. Parkinson disease); however, the changes are only in the upper cortex and not diffuse.[42]
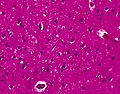
CJD. (WC/DRdoubleB)
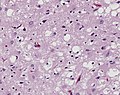
Spongiform changes in CJD. (CDC/ Teresa Hammett)
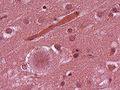
Florid plaques in vCJD. (WC/Sbrandner)
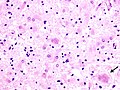
Florid plaques in vCJD - low mag. (WC/CDC.gov)
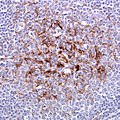
vCJD - prion protein immunostain tonsil. (WC/Sbrandner)
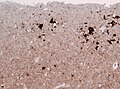
sCJD - prion protein immunostain cortex. (WC/jensflorian)

sCJD - prion protein immunostain cerebellum. (WC/jensflorian)

,_H%26E.jpg)





Alpha-synucleinopathies
Without clincial information Parkinson's disease and Dementia with Lewy bodies cannot separated in histology.
Dementia with Lewy bodies
General
Clinical features:
- Parkinsonian features.
- Hallucinations (visual).
- Progressive cognitive decline with fluctuations.
Microscopic
Features:
- Lewy bodies.
- Lewy neurites.
Note: Cortical Lewy bodies are easily missed in HE.
IHC
- Alpha-synuclein +ve.
Images

Lewy bodies in frontal cortex of DLB. (WC/jensflorian)
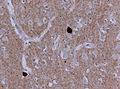
Alpha-synculein IHC showing cortical Lewy bodies. (WC/jensflorian)
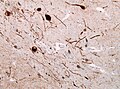
Alpha-synculein IHC showing Lewy Neurites in DLB. (WC/jensflorian)



Parkinson disease
General
- Common - often sporadic.
- May be genetic.
Clinical TRAP:[43]
- Tremor.
- Rigidity.
- Akinesia.
- Postural instability.
Genetics:[44]
Gross
Features:[47]
- Abnormally pale substantia nigra.
- Pigmentation increases with age.
- Pale locus ceruleus.
Notes:
- Substantia nigra is a midbrain structure.
- Image: Midbrain - schematic (WC).
Microscopic
Features:[47]
- Loss of pigmented (catecholaminergic) neurons in the substantia nigra and locus ceruleus.
- Gliosis - due to neuron loss.
- Lewy bodies (in remaining neurons) - key feature.
- Eosinophilic cytoplasmic inclusion with "dense" (darker) core and pale (surrounding) halo.
- Consist of filaments composed of alpha-synuclein.
- Eosinophilic cytoplasmic inclusion with "dense" (darker) core and pale (surrounding) halo.
- Lewy neurites - alpha-synuclein positive processes.
IHC
- Alpha-synuclein +ve.
Images

Schematic progression of PD (PLOSone/Jubault et al.).
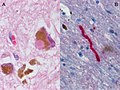
Lewy body (HE, left) and Lewy neurite (aSyn IHC, right).
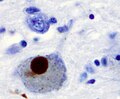
Alpha-synculein positive Lewy body (WC/Marvin101).
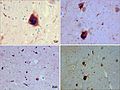
Lewy body and Lewy neurites (WC/Suraj Rajan).



.jpg)
Molecular
- Hereditary forms in less than 10% of the cases
- Involved genes are consecutively labeled PARK1, PARK2....
Multiple system atrophy
Multiple system atrophy is a neurodegenerative disease of the parkinsonism-plus disorder group.
General
Clinical findings variable:
- Parkinsonism (stiatonigral degeneration, MSA-P).
- Ataxia (olivo-ponto-cerebellar degeneration, MSA-C).
- Autonomic dysfunction (Shy-Drager syndrome, depreceated).
- Clinical onset between 40-60 years.
- Progedient tremor, atxia, laryngeal paresis, wakness, cognitive decline.
- Patients usually succumb after 6 years from aspiration pneumonia.
DDx:
- Spinocerebellar ataxia.
- Parkinson disease.
- Motor-neuron disease.
- Lewy-Body disease.
Macroscopy
- Cerebral (mild) & cerebellar atrophy.
- greenish putamen.
- Discoloration Substantia nigra and Locus coeruleus
Microscopic
Features:
- Inclusions cerebral, subcortical white matter, cerebellar.
- Neuronal loss and gliosis (absent in minimal-change MSA).
- Alpha-synuclein-rich glial and neuronal cytoplasmic inclusions in white matter (finding at autopsy).[48]
- Pons and Putamen:
- Nuclear inclusions (sparse in most cases).
- Neuropil threads (alpha-synuclein).
- Loss of myelinated fibers from external capsule, striatum and pallidum.
Images

Gray-brown discoloration in putamen of a striatonigral-type MSA (WC/jensflorian).
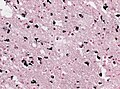
Gallays silver stain showing MSA-typic inclusions (WC/jensflorian).

a-synuclein IHC showing Glial and neuronal cytoplasmic inclusion in the pons of a MSA case (WC/jensflorian).



Molecular
- No known alpha-synuclein mutation.
- Genetic variants of SNCA gene assoicated with MSA. [51]
Tauopathies
More than 20 different degenerative disorders can be classified as tauopathies.[52] FTLD-tau is an umbrella term used for tauopathies including PSP, CBD, PiD and GGT. [53]
Argyrophilic grain disease
Corticobasal degeneration
- AKA CBD.
- Symptoms may vary:
- Progressive asymmetrical rigidity and apraxia, progressive aphasia or dementia.
- Neuronal and glial Tau-positive inclusions.[54]
- Astrocytic plaques.
- Thread-like lesions and coiled bodies.
- Ballooned neurons +/-.
- Pathology is cortical and striatal and Gallyas-positive.
- Neuronal loss in the substantia nigra.
DD: PSP (widespread neurofibrillary degeneration, with characteristic globose NFT).
Globular glial tauopathies
- Commonly abbreviated GGT.
- AKA sporadic multiple system tauopathy.
- Rare disease.[55]
- Combination of frontotemporal dementia and motor neuron disease or only part thereof.
- 4-repeat tauopathy.
Microscopic
- Globular oligodendroglial and astrocytic Tau inclusions.
- Absence of tufted astrocytes.
- Mostly Gallyas-negative.
Progressive supranuclear palsy
- Commonly abbreviated PSP.
- AKA Steele-Richardson-Olszewski syndrome.
General
- Diagnosis - clinical.[56]
Clinical:
- Impaired control of gaze, esp. difficulty looking up and down (supranuclear palsy).[57]
- Parkinsonism.[11]
Microscopic
- Globose neurofibrillary tangles in neurons.
- Coiled bodies in oligodendrocytes.
- Wire coil-like structure around the nucleus.
- Tufted astrocytes.
- Near impossible to see without IHC - specifically AT8.
- Cellular processes filled with crap.
- Star-like appearance; looks like a road network where all the roads lead to one place (Parisian star).
- Grumose degeneration of the cerebellar dentate nucleus.
Images:
Pick disease
General
- Dementia.
Gross
Microscopic
Features:[61]
- Pick cells = large ballooned neurons.
- Pick bodies = round, homogenous, intracytoplasmic inclusions, size ~10 micrometers.
Image(s):
TDP Proteinopathies
FTLD-FET
- Clinical manifestations depend on the distribution of the pathologic alterations in the CNS
- Currently 3 disorders among the FTLD-FET subgroup.
- In contrast to ALS-FUS, no genetic alterations of FUS have been reported to date for cases within the FTLD-FUS group.
- 5–10% of all FTLD cases
- Deposited Proteins: FUS, EWS, TAF-15.
- FUS‐positive inclusions in FTLD cases show co‐aggregation of TAF15 and EWS
- (Different from ALS-FUS)
DDx (also FUS+ve):
- Spinocerebellar Ataxia (SCA)
- Huntington Disease (SD)
Atypical FTLD‐U
- Early onset frontotemporal dementia, rapidly progressive psycho‐behavioural changes.
- Neuronal cytoplasmic inclusions in hippocampus and frontotemporal lobes.
- Ubiquitin+ve, tau/TDP‐ve.
- FET+ve inclusions
- Unique vermiform filamentous neuronal nuclear inclusions.
- Caudate nucleus head degeneration and hippocampal sclerosis.
Basophilic inclusion body disease
- AKA: BIBD.
- Variable clinic (behavioral, cognitive alterations, parkinsonism, motor neuron diseases, ALS-like).
- Age of onset: 35-70 years.
- Intraneuronal cytoplasmic basophilic inclusion bodies.
- FUS+ve (universally).
- TAF15+ve
- alpha-Internexin+ve.
Neuronal Intermediate Filament Inclusion Disease
- AKA: NIFID.
- Hyaline conglomerates (brightly eosinophilic branching fibrillar structures embedded in a round, well-delineated, glassy vacuole).
- FUS+ve (heterogenous).
- Ubiquitin +/-ve.
- NF +ve (some subunits).
- p62 +/-ve.
- TDP43-ve.
- Tau-ve.
- α-synuclein-ve.
Other
Chronic traumatic encephalopathy
- Abbreviated CTE.
Huntington disease
General
- Autosomal dominant inheritance.
- Mutation in Huntington gene (HTT):[64]
- 11-34 CAG repeat = normal.[65]
- >42 CAG repeat = Huntington disease.
Clinical:[66]
- Early onset dementia.
- Involuntary movements (chorea) - both arms and legs.
- Behaviour changes, e.g. grimacing.
- Speech changes.
Gross
Note:
- A normal caudate bulges into the ventricle.
Images:
Microscopic
Features:[66]
- Neuron loss.
- Gliosis.
Binswanger disease
General
- Multi-infarct dementia affecting subcortical white matter.
- Waste-basket diagnosis; diagnosed if CADASIL and amyloidosis have been excluded.
- Diagnosis has been controversial -- most with this entity (in the past) were diagnosed with Alzheimer's disease.
Microscopic
Features:
- Subcortical lesions that replace the myelin consisting of macrophages.
Frontotemporal lobar degeneration with ubiquitinated inclusions
Abbreviated FTLD with ubiquitinated inclusions or FTLD-TDP43.
General
- There are several forms of frontotemporal dementia.
- Related to amyotrophic lateral sclerosis (ALS); also a TDP-43 pathology.[69]
- There are several subtypes of FTLD with TDP-43.
Gross
- Frontal and temporal lobe atrophy.
Image:
Amyotrophic lateral sclerosis
- Abbreviated ALS.
General
- AKA Lou Gehrig's disease.
- Characterized by motor neuron death.
- May be familial and associated with C9orf72 expansion, or SOD1, FUS and TARDBP mutations.[70][71]
- Pathological protein aggregates cause dysfunction of RNA-binding proteins.
Clinical
- Peak incidence: 50-60yrs.
- 2-5 per 100,000 individuals worldwide.
- Dead after disease onset: Usu. 2-5yrs.
- Weakness (Progressive bulbar, limb, thoracic, and abdominal muscle atrophy).
- About 20% of ALS cases develop frontotemporal lobar degeneration (FTLD).
- Environmental toxins are discussed (Guam ALS).[72]
Microscopic
- Loss of the giant cells of Betz.
- Motor neurons with eosinophilic inclusions (Bunina bodies).
- PAS positive cytoplasmic inclusions.
- Motor neuron loss + reactive gliosis + neurogenic muscular atrophy.
- Loss of myelinated axons in the lateral and anterior columns of the spinal cord.
- Ubiquitinated cytoplasmic inclusions.[74]
- TDP-43 proteinopathy in motor neurons (90% of all sporadic ALS cases).
- C9orf72 expansion cases: p62+ve, TDP-43-ve inclusions in the dentate gyrus, neocortex, and cerebellum.[76]
Images:
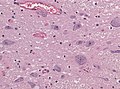
Motor neuron loss in ALS-TDP. (WC)

pTDP-43 positive inclusions in neurons. (WC)


![[1]](http://en.wikipedia.org/wiki/File:Cerebral_amyloid_angiopathy_-2a-_amyloid_beta_-_high_mag.jpg){kind=link}
![[2]](http://en.wikipedia.org/wiki/File:SpongiformChangeCJD.jpg){kind=link}
![[3]](http://firstaidteam.com/usmlerximages/v/USMLERxLewy+bodies.gif.html){kind=link}
![[4]](https://de.wikipedia.org/wiki/Datei:LewyBodies_small.JPG){kind=link}
![[6]](https://commons.wikimedia.org/wiki/File:DLB_frontal_lewy_bodies_HE.jpg){kind=link}
![[7]](https://commons.wikimedia.org/wiki/File:MSA_Gallays_Papp_Lantos_inclusions.jpg){kind=link}
![[8]](https://commons.wikimedia.org/wiki/File:Als_inclusions.png){kind=link}
![[9]](https://commons.wikimedia.org/wiki/File:FTLD_TSP43_hippocampus.jpg){kind=link}
![[10]](http://brain.oxfordjournals.org/content/brain/134/9/2595/F1.medium.gif){kind=link}
![[11]](https://commons.wikimedia.org/wiki/File:PSP_Tau_tufted_astrocyte.jpg){kind=link}
![[12]](http://frontalcortex.com/gallery/pics/gliageek_PBtau.jpg){kind=link}
![[13]](http://webeye.ophth.uiowa.edu/eyeforum/cases-i/case155/4-Progressive-Supranuclear-Palsy-histology.jpg){kind=link}
![[14]](http://de.wikipedia.org/wiki/Silberkornkrankheit#/media/File:AGD_gallays.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DDx:
- Spinal muscular atrophy.
- Primary Lateral Sclerosis.
- Hereditary Spastic Paraparesis (HSP).
Hallervorden-Spatz disease
- AKA pantothenate kinase-associated neurodegeneration.
General
- Uncommon.
Microscopic
Features:[79]
- Axonal spheroids.
- Iron deposition.
Images:
Stains
- Prussian blue +ve.
See also
References
- ↑ 1.0 1.1 1.2 1.3 Dickson DW (2009). "Neuropathology of non-Alzheimer degenerative disorders". Int J Clin Exp Pathol 3 (1): 1–23. PMC 2776269. PMID 19918325. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2776269/?tool=pubmed.
- ↑ Uversky, VN. (Oct 2008). "Alpha-synuclein misfolding and neurodegenerative diseases.". Curr Protein Pept Sci 9 (5): 507-40. PMID 18855701.
- ↑ MUN. 15 November 2010.
- ↑ 4.0 4.1 Seelaar H, Klijnsma KY, de Koning I, et al. (May 2010). "Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration". J. Neurol. 257 (5): 747–53. doi:10.1007/s00415-009-5404-z. PMC 2864899. PMID 19946779. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2864899/.
- ↑ Kumaran R, Kingsbury A, Coulter I, et al. (October 2007). "DJ-1 (PARK7) is associated with 3R and 4R tau neuronal and glial inclusions in neurodegenerative disorders". Neurobiol. Dis. 28 (1): 122–32. doi:10.1016/j.nbd.2007.07.012. PMID 17719794.
- ↑ Geser F, Brandmeir NJ, Kwong LK, et al. (May 2008). "Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis". Arch. Neurol. 65 (5): 636–41. doi:10.1001/archneur.65.5.636. PMID 18474740.
- ↑ URL: http://dictionary.reference.com/browse/skein. Accessed on: 20 November 2010.
- ↑ Shiau, Carolyn; Toren, Andrew (2006). Toronto Notes 2006: Comprehensive Medical Reference (Review for MCCQE 1 and USMLE Step 2) (22nd edition (2006) ed.). Toronto Notes for Medical Students, Inc.. pp. PS19. ISBN 978-0968592861.
- ↑ Tuite, PJ.; Krawczewski, K. (Apr 2007). "Parkinsonism: a review-of-systems approach to diagnosis.". Semin Neurol 27 (2): 113-22. doi:10.1055/s-2007-971174. PMID 17390256.
- ↑ Ahmed, Z.; Asi, YT.; Sailer, A.; Lees, AJ.; Houlden, H.; Revesz, T.; Holton, JL. (Nov 2011). "Review: The neuropathology, pathophysiology and genetics of multiple system atrophy.". Neuropathol Appl Neurobiol. doi:10.1111/j.1365-2990.2011.01234.x. PMID 22074330.
- ↑ 11.0 11.1 Bertram, K.; Williams, DR. (Apr 2012). "Visual hallucinations in the differential diagnosis of parkinsonism.". J Neurol Neurosurg Psychiatry 83 (4): 448-52. doi:10.1136/jnnp-2011-300980. PMID 22228724.
- ↑ Mahmoud, F.; Tampi, RR. (Oct 2011). "Valproic Acid-Induced Parkinsonism in the Elderly: A Comprehensive Review of the Literature.". Am J Geriatr Pharmacother. doi:10.1016/j.amjopharm.2011.09.002. PMID 21993183.
- ↑ Gerlach, M.; Riederer, P.; Przuntek, H.; Youdim, MB. (Dec 1991). "MPTP mechanisms of neurotoxicity and their implications for Parkinson's disease.". Eur J Pharmacol 208 (4): 273-86. PMID 1815982.
- ↑ Korczyn, AD. (Apr 2015). "Vascular parkinsonism-characteristics, pathogenesis and treatment.". Nat Rev Neurol. doi:10.1038/nrneurol.2015.61. PMID 25917706.
- ↑ Vilensky, JA.; Gilman, S.; McCall, S. (Jul 2010). "A historical analysis of the relationship between encephalitis lethargica and postencephalitic parkinsonism: a complex rather than a direct relationship.". Mov Disord 25 (9): 1116-23. doi:10.1002/mds.22908. PMID 20629120.
- ↑ Chauhan, NB. (2014). "Chronic neurodegenerative consequences of traumatic brain injury.". Restor Neurol Neurosci 32 (2): 337-65. doi:10.3233/RNN-130354. PMID 24398724.
- ↑ Nelson, PT.; Alafuzoff, I.; Bigio, EH.; Bouras, C.; Braak, H.; Cairns, NJ.; Castellani, RJ.; Crain, BJ. et al. (May 2012). "Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature.". J Neuropathol Exp Neurol 71 (5): 362-81. doi:10.1097/NEN.0b013e31825018f7. PMID 22487856.
- ↑ Duyckaerts, C.; Sazdovitch, V.; Ando, K.; Seilhean, D.; Privat, N.; Yilmaz, Z.; Peckeu, L.; Amar, E. et al. (Feb 2018). "Neuropathology of iatrogenic Creutzfeldt-Jakob disease and immunoassay of French cadaver-sourced growth hormone batches suggest possible transmission of tauopathy and long incubation periods for the transmission of Abeta pathology.". Acta Neuropathol 135 (2): 201-212. doi:10.1007/s00401-017-1791-x. PMID 29209767.
- ↑ Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, JD.; Nicoll, AJ.; Kenny, J.; Launchbury, F.; Linehan, J. et al. (Sep 2015). "Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy.". Nature 525 (7568): 247-50. doi:10.1038/nature15369. PMID 26354483.
- ↑ Mitchell, Richard; Kumar, Vinay; Fausto, Nelson; Abbas, Abul K.; Aster, Jon (2011). Pocket Companion to Robbins & Cotran Pathologic Basis of Disease (8th ed.). Elsevier Saunders. pp. 674-5. ISBN 978-1416054542.
- ↑ Nieuwenhuis-Mark, RE.. "Diagnosing Alzheimer's dementia in Down syndrome: problems and possible solutions.". Res Dev Disabil 30 (5): 827-38. doi:10.1016/j.ridd.2009.01.010. PMID 19269132.
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 104311
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 600759
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 107741
- ↑ Braak H, Braak E, Bohl J (1993). "Staging of Alzheimer-related cortical destruction". Eur. Neurol. 33 (6): 403–8. PMID 8307060.
- ↑ Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Aster, Jon (2009). Robbins and Cotran pathologic basis of disease (8th ed.). Elsevier Saunders. pp. 1317. ISBN 978-1416031215.
- ↑ Braak, H.; Braak, E. (1991). "Neuropathological stageing of Alzheimer-related changes.". Acta Neuropathol 82 (4): 239-59. PMID 1759558.
- ↑ URL: http://www.pakmed.net/academic/age/alz/alz030.htm. Accessed on: 12 November 2010.
- ↑ URL: http://faculty.washington.edu/alexbert/MEDEX/Fall/NeuroPath_Obj.htm. Accessed on: 13 November 2010.
- ↑ Mirra, SS.; Heyman, A.; McKeel, D.; Sumi, SM.; Crain, BJ.; Brownlee, LM.; Vogel, FS.; Hughes, JP. et al. (Apr 1991). "The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease.". Neurology 41 (4): 479-86. PMID 2011243.
- ↑ URL: http://library.med.utah.edu/WebPath/EXAM/IMGQUIZ/npfrm.html. Accessed on: 5 December 2010.
- ↑ Montine, TJ.; Phelps, CH.; Beach, TG.; Bigio, EH.; Cairns, NJ.; Dickson, DW.; Duyckaerts, C.; Frosch, MP. et al. (Jan 2012). "National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach.". Acta Neuropathol 123 (1): 1-11. doi:10.1007/s00401-011-0910-3. PMID 22101365.
- ↑ Thal, DR.; Rüb, U.; Orantes, M.; Braak, H. (Jun 2002). "Phases of A beta-deposition in the human brain and its relevance for the development of AD.". Neurology 58 (12): 1791-800. PMID 12084879.
- ↑ Braak, H.; Braak, E. (1991). "Neuropathological stageing of Alzheimer-related changes.". Acta Neuropathol 82 (4): 239-59. PMID 1759558.
- ↑ Mirra, SS.; Heyman, A.; McKeel, D.; Sumi, SM.; Crain, BJ.; Brownlee, LM.; Vogel, FS.; Hughes, JP. et al. (Apr 1991). "The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease.". Neurology 41 (4): 479-86. PMID 2011243.
- ↑ 36.0 36.1 Watts JC, Balachandran A, Westaway D (March 2006). "The expanding universe of prion diseases". PLoS Pathog. 2 (3): e26. doi:10.1371/journal.ppat.0020026. PMC 1434791. PMID 16609731. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1434791/.
- ↑ 37.0 37.1 Mitchell, Richard; Kumar, Vinay; Fausto, Nelson; Abbas, Abul K.; Aster, Jon (2011). Pocket Companion to Robbins & Cotran Pathologic Basis of Disease (8th ed.). Elsevier Saunders. pp. 672. ISBN 978-1416054542.
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 600072
- ↑ Mitchell, Richard; Kumar, Vinay; Fausto, Nelson; Abbas, Abul K.; Aster, Jon (2011). Pocket Companion to Robbins & Cotran Pathologic Basis of Disease (8th ed.). Elsevier Saunders. pp. 671. ISBN 978-1416054542.
- ↑ Burton, Julian L.; Rutty, Guy N. (2010). The Hospital Autopsy A Manual of Fundamental Autopsy Practice (3rd ed.). Oxford University Press. pp. 83. ISBN 978-0340965146.
- ↑ URL: http://moon.ouhsc.edu/kfung/jty1/opaq/PathQuiz/N0I002-PQ01-M.htm. Accessed on: 19 October 2010.
- ↑ Lefkowitch, Jay H. (2006). Anatomic Pathology Board Review (1st ed.). Saunders. pp. 419 Q4. ISBN 978-1416025887.
- ↑ URL: http://www.nysslha.org/i4a/pages/index.cfm?pageid=3519. Accessed on: 30 March 2011.
- ↑ Mitchell, Richard; Kumar, Vinay; Fausto, Nelson; Abbas, Abul K.; Aster, Jon (2011). Pocket Companion to Robbins & Cotran Pathologic Basis of Disease (8th ed.). Elsevier Saunders. pp. 677. ISBN 978-1416054542.
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 609007
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 602544
- ↑ 47.0 47.1 Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Aster, Jon (2009). Robbins and Cotran pathologic basis of disease (8th ed.). Elsevier Saunders. pp. 1319. ISBN 978-1416031215.
- ↑ Wenning, GK.; Stefanova, N.; Jellinger, KA.; Poewe, W.; Schlossmacher, MG. (Sep 2008). "Multiple system atrophy: a primary oligodendrogliopathy.". Ann Neurol 64 (3): 239-46. doi:10.1002/ana.21465. PMID 18825660.
- ↑ MUN. 16 November 2010.
- ↑ "Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy". Neuropathol. Appl. Neurobiol. 33 (6): 615–20. 2007. doi:10.1111/j.1365-2990.2007.00907.x. PMID 17990994.
- ↑ Pimenta, PF.; da Silva, RP.; Sacks, DL.; da Silva, PP. (Apr 1989). "Cell surface nanoanatomy of Leishmania major as revealed by fracture-flip. A surface meshwork of 44 nm fusiform filaments identifies infective developmental stage promastigotes.". Eur J Cell Biol 48 (2): 180-90. PMID 2743996.
- ↑ Williams, DR. (Oct 2006). "Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau.". Intern Med J 36 (10): 652-60. doi:10.1111/j.1445-5994.2006.01153.x. PMID 16958643.
- ↑ Forrest, SL.; Kril, JJ.; Stevens, CH.; Kwok, JB.; Hallupp, M.; Kim, WS.; Huang, Y.; McGinley, CV. et al. (Feb 2018). "Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies.". Brain 141 (2): 521-534. doi:10.1093/brain/awx328. PMID 29253099.
- ↑ Dickson, DW.; Bergeron, C.; Chin, SS.; Duyckaerts, C.; Horoupian, D.; Ikeda, K.; Jellinger, K.; Lantos, PL. et al. (Nov 2002). "Office of Rare Diseases neuropathologic criteria for corticobasal degeneration.". J Neuropathol Exp Neurol 61 (11): 935-46. PMID 12430710.
- ↑ Ahmed, Z.; Bigio, EH.; Budka, H.; Dickson, DW.; Ferrer, I.; Ghetti, B.; Giaccone, G.; Hatanpaa, KJ. et al. (Oct 2013). "Globular glial tauopathies (GGT): consensus recommendations.". Acta Neuropathol 126 (4): 537-544. doi:10.1007/s00401-013-1171-0. PMID 23995422.
- ↑ 56.0 56.1 URL: http://emedicine.medscape.com/article/1151430-overview. Accessed on: 11 November 2010.
- ↑ Levy, R. (Jun 2011). "[Progressive supranuclear palsy: what's new?].". Geriatr Psychol Neuropsychiatr Vieil 9 (2): 191-201. doi:10.1684/pnv.2011.0271. PMID 21690028.
- ↑ Williams DR, Lees AJ (March 2009). "Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges". Lancet Neurol 8 (3): 270–9. doi:10.1016/S1474-4422(09)70042-0. PMID 19233037.
- ↑ URL: http://neuropathologyblog.blogspot.com/2008/03/grumose-degeneration-in-cerebellar.html. Accessed on: 4 December 2010.
- ↑ Yamanouchi H, Yokoo H, Yuhara Y, et al. (March 2002). "An autopsy case of ornithine transcarbamylase deficiency". Brain Dev. 24 (2): 91–4. PMID 11891099.
- ↑ 61.0 61.1 Mitchell, Richard; Kumar, Vinay; Fausto, Nelson; Abbas, Abul K.; Aster, Jon (2011). Pocket Companion to Robbins & Cotran Pathologic Basis of Disease (8th ed.). Elsevier Saunders. pp. 676. ISBN 978-1416054542.
- ↑ URL: http://medical-dictionary.thefreedictionary.com/Walnut+Brain. Accessed on: 14 March 2012.
- ↑ Grossman, M. (Feb 2010). "Primary progressive aphasia: clinicopathological correlations.". Nat Rev Neurol 6 (2): 88-97. doi:10.1038/nrneurol.2009.216. PMID 20139998.
- ↑ Kumar P, Kalonia H, Kumar A (2010). "Huntington's disease: pathogenesis to animal models". Pharmacol Rep 62 (1): 1–14. PMID 20360611.
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 613004
- ↑ 66.0 66.1 Lefkowitch, Jay H. (2006). Anatomic Pathology Board Review (1st ed.). Saunders. pp. 415 Q44. ISBN 978-1416025887.
- ↑ URL: http://moon.ouhsc.edu/kfung/jty1/NeuroTest/Q07-Ans.htm. Accessed on: 29 October 2010.
- ↑ URL: http://path.upmc.edu/cases/case117/gross.html. Accessed on: 3 January 2012.
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 105400
- ↑ 70.0 70.1 Mitchell, Richard; Kumar, Vinay; Fausto, Nelson; Abbas, Abul K.; Aster, Jon (2011). Pocket Companion to Robbins & Cotran Pathologic Basis of Disease (8th ed.). Elsevier Saunders. pp. 679. ISBN 978-1416054542.
- ↑ Guerrero, EN.; Wang, H.; Mitra, J.; Hegde, PM.; Stowell, SE.; Liachko, NF.; Kraemer, BC.; Garruto, RM. et al. "TDP-43/FUS in motor neuron disease: Complexity and challenges.". Prog Neurobiol 145-146: 78-97. doi:10.1016/j.pneurobio.2016.09.004. PMID 27693252.
- ↑ Chernoff, N.; Hill, DJ.; Diggs, DL.; Faison, BD.; Francis, BM.; Lang, JR.; Larue, MM.; Le, TT. et al. (2017). "A critical review of the postulated role of the non-essential amino acid, β-N-methylamino-L-alanine, in neurodegenerative disease in humans.". J Toxicol Environ Health B Crit Rev 20 (4): 1-47. doi:10.1080/10937404.2017.1297592. PMID 28598725.
- ↑ Saberi, S.; Stauffer, JE.; Schulte, DJ.; Ravits, J. (Nov 2015). "Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants.". Neurol Clin 33 (4): 855-76. doi:10.1016/j.ncl.2015.07.012. PMID 26515626.
- ↑ Leigh, PN.; Anderton, BH.; Dodson, A.; Gallo, JM.; Swash, M.; Power, DM. (Nov 1988). "Ubiquitin deposits in anterior horn cells in motor neurone disease.". Neurosci Lett 93 (2-3): 197-203. PMID 2853844.
- ↑ Nakamura, S.; Wate, R.; Kaneko, S.; Ito, H.; Oki, M.; Tsuge, A.; Nagashima, M.; Asayama, S. et al. (Feb 2014). "An autopsy case of sporadic amyotrophic lateral sclerosis associated with the I113T SOD1 mutation.". Neuropathology 34 (1): 58-63. doi:10.1111/neup.12049. PMID 23773010.
- ↑ Al-Sarraj, S.; King, A.; Troakes, C.; Smith, B.; Maekawa, S.; Bodi, I.; Rogelj, B.; Al-Chalabi, A. et al. (Dec 2011). "p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS.". Acta Neuropathol 122 (6): 691-702. doi:10.1007/s00401-011-0911-2. PMID 22101323.
- ↑ Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, KJ.; Nishimura, AL.; Sreedharan, J.; Hu, X.; Smith, B. et al. (Feb 2009). "Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6.". Science 323 (5918): 1208-1211. doi:10.1126/science.1165942. PMID 19251628.
- ↑ URL: http://pathology.mc.duke.edu/neuropath/CNSlecture4/CNSlecture4.htm. Accessed on: 30 August 2011.
- ↑ URL: http://path.upmc.edu/cases/case207/dx.html. Accessed on: 11 January 2012.