Difference between revisions of "Langerhans cell histiocytosis"
Jump to navigation
Jump to search
| Line 2: | Line 2: | ||
It has been referred to by several eponyms - '''Hand-Schüller-Christian disease''', '''Abt-Letterer-Siwe disease''' or '''Letterer-Siwe disease''', and '''histiocytosis X'''. | It has been referred to by several eponyms - '''Hand-Schüller-Christian disease''', '''Abt-Letterer-Siwe disease''' or '''Letterer-Siwe disease''', and '''histiocytosis X'''. | ||
This article deals with LCH in general. A separate article exists for ''[[pulmonary Langerhans cell histiocytosis]]''. | |||
==General== | ==General== | ||
Revision as of 06:25, 23 December 2015
Langerhans cell histiocytosis, abbreviated LCH, is a rare disorder of tissue macrophages. It broadly fits into the category of histiocytoses. It used to known as eosinophilic granuloma.
It has been referred to by several eponyms - Hand-Schüller-Christian disease, Abt-Letterer-Siwe disease or Letterer-Siwe disease, and histiocytosis X.
This article deals with LCH in general. A separate article exists for pulmonary Langerhans cell histiocytosis.
General
Overview
LCH is really four (or three) diseases (depending on how one classifies it) - that happen to share the same histology:[1][2]
| Disease | Other name(s) | Prognosis | Demographic | Location | Risks/cause |
|---|---|---|---|---|---|
| Pulmonary Langerhans cell histiocytosis | Eosinophilic granuloma | good with smoking cessation | adults - smokers | lung only; typically upper lung field | due to smoking |
| Multifocal multisystem Langerhans cell histiocytosis | multisystem LCH, Letterer-Siwe disease | outcome dependent on organ involved,[3] natural history 2 year survival, 50% five year survival with treatment | usu. children < 2 years old, rarely adults[4] | multiple systems (skin, spleen, liver, lung, bone marrow) | possibly genetic ‡ |
| Unifocal Langerhans cell histiocytosis † | Eosinophilic granuloma | may spontaneously regress, may cure with surgery | children (?) | bone only | possibly genetic ‡ |
| Multifocal unisystem Langerhans cell histiocytosis † | multifocal LCH, eosinophilic granuloma, Hand-Schuller-Christian syndrome = bone defect, diabetes insipidus & exopthalmos | may spontaneously regress, may cure with surgery (?) | children (?) | usu. bone; may be in: skin, lungs, stomach | possibly genetic ‡ |
Note:
- † Robbins lumps these groups together.
- ‡ Incompletely understood. Somatic BRAF mutations identified in approximately half of the individuals.[5][6]
Clinical presentation
Features - dependent on subtype:[1]
- May present with fever, anemia, bone pain, bone fracture, diabetes insipidus, exophthalmos.
- Can be an incidental finding.
Microscopic
Features:[2]
- Langerhans cells histiocytes - key feature.
- Clusters of cells (histiocytes) with a reniform (kidney-shaped) nucleus and abundant foamy cytoplasm.
- Nucleus may look like a "coffee bean", i.e. have nuclear grooves (similar to those in papillary thyroid carcinoma) -- appearance dependent on the rotation of the nucleus.[7] May be called "buttock cells".
- Chromatin pattern: fine granular, light gray.
- Clusters of cells (histiocytes) with a reniform (kidney-shaped) nucleus and abundant foamy cytoplasm.
- +/-Eosinophils - often prominent.
- +/-Fibrosis - common.
- +/-Other inflammatory cells - neutrophils, plasma cells (uncommon).
- +/-Multinucleated giant cells - uncommon.
DDx:
- Kimura disease - eosinophilia.
- See lymph node pathology.
- See lesions with many eosinophils.
Images
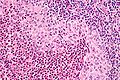
LCH - lymph node - very high mag. (WC)
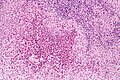
LCH - lymph node - high mag. (WC)


www:
IHC
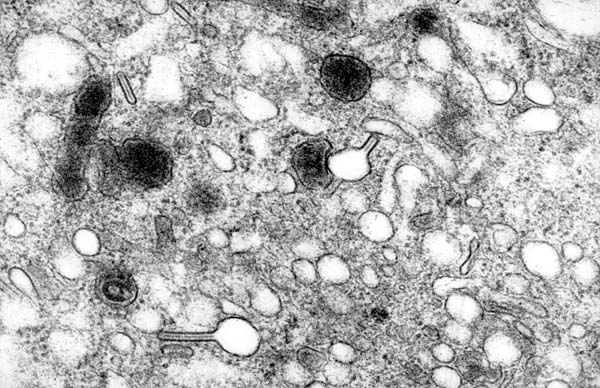
Electron microscopy
Etiology:
- Cell membrane invagination.[9]
Appearance:
- Electron dense, cytoplasmic tennis racket-like body.
Images:
{kind=link}
See also
References
- ↑ 1.0 1.1 Mitchell, Richard; Kumar, Vinay; Fausto, Nelson; Abbas, Abul K.; Aster, Jon (2011). Pocket Companion to Robbins & Cotran Pathologic Basis of Disease (8th ed.). Elsevier Saunders. pp. 338-9. ISBN 978-1416054542.
- ↑ 2.0 2.1 Chhabra, UD.; Desai, SS.; Jambhekar, NA. (Jul 2004). "Langerhans' cell histiocytosis: a clinicopathological study of 50 cases.". Indian J Pathol Microbiol 47 (3): 370-6. PMID 16295427.
- ↑ Minkov, M. (Apr 2011). "Multisystem Langerhans cell histiocytosis in children: current treatment and future directions.". Paediatr Drugs 13 (2): 75-86. doi:10.2165/11538540-000000000-00000. PMID 21351807.
- ↑ Garg, A.; Kumar, P. (Jan 2012). "Multisystem Langerhans cell histiocytosis in adult.". Indian J Dermatol 57 (1): 58-60. doi:10.4103/0019-5154.92683. PMID 22470214.
- ↑ Badalian-Very, G.; Vergilio, JA.; Degar, BA.; MacConaill, LE.; Brandner, B.; Calicchio, ML.; Kuo, FC.; Ligon, AH. et al. (Sep 2010). "Recurrent BRAF mutations in Langerhans cell histiocytosis.". Blood 116 (11): 1919-23. doi:10.1182/blood-2010-04-279083. PMID 20519626.
- ↑ Badalian-Very, G.; Vergilio, JA.; Degar, BA.; Rodriguez-Galindo, C.; Rollins, BJ. (Jan 2012). "Recent advances in the understanding of Langerhans cell histiocytosis.". Br J Haematol 156 (2): 163-72. doi:10.1111/j.1365-2141.2011.08915.x. PMID 22017623.
- ↑ BN. 15 March 2011.
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 604862
- ↑ URL: http://path.upmc.edu/cases/case147/micro.html. Accessed on: 7 January 2012.
- ↑ URL: http://path.upmc.edu/cases/case298.html. Accessed on: 14 January 2012.