Glycogen storage diseases
Glycogen storage diseases a group of diseases characterized by the accumulation of glycogen.
Clinical picture
- Exercise intolerance
- Usually due to specific muscle enzyme defects
DDx:
- Mitochondriopathies
- Carnitine palmitoyltransferase II (CPT2) deficiency
General microscopic
Features:[1]
- +/-Vacuolated muscle fibres.
- acid phosphatase+ve in vaculoes.
- PAS+ve.
Images:
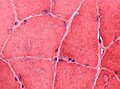
Abnormal glycogen is not easy to spot in this muscle biopsy HE stain (WC/jensflorian)
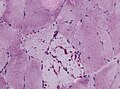
Intramuscular glycogen is usually PAS+++ve (WC/jensflorian)

Lack of staining in intramuscular deposits, Trichrom Gömöri (WC/jensflorian)
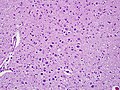
Lafora-like polyglucosan bodies in the CNS, low magnification (WC/jensflorian)
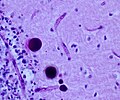
Lafora-like polyglucosan bodies in the CNS, higher magnification(WC/marvin101)
Large vacuoles in the liver, HE stain (WC/Netha Hussain)





Electron microscopy
- Electron dense deposits.
Specific diseases
Pompe disease
General
Deficiency of alpha-1,4-glucosidase; it degrades glycogen to glucose in lysosomes.
Clinical:
- Floppy baby.
- Big heart.
- Often early death from cardiac failure.
- Pompe vacuoles.jpg
Large vacuoles in Pompe disease (H&E, WC/jensflorian)
- Phenotypical-variation-within-22-families-with-Pompe-disease-1750-1172-8-182-S1.ogv
Clinical phenotype in Pompe disease (WC/Wens et. al.)
Cori disease
General
- Hepatomegaly.
Microscopic
Features:
- Hypertrophic hepatocytes with pale cytoplasm.
- Classically: PAS +ve, PAS-D -ve.
- Portal fibrosis.
Image:
{kind=link}
Stains
See also
References
- ↑ URL: http://neuromuscular.wustl.edu/pathol/acidmchi.htm. Accessed on: 11 January 2011.
- ↑ URL: http://www.ncbi.nlm.nih.gov/omim/606800. Accessed on: 11 January 2011.
- ↑ URL: http://www.ncbi.nlm.nih.gov/omim/232400. Accessed on: 25 January 2011.