Difference between revisions of "Medulloblastoma"
Jensflorian (talk | contribs) (→Histologically defined: desmoplastic) |
Jensflorian (talk | contribs) |
||
| (6 intermediate revisions by the same user not shown) | |||
| Line 1: | Line 1: | ||
'''Medulloblastoma''' is a [[malignant]] [[small round cell tumour]] that is found in the [[cerebellum]]. | {{ Infobox diagnosis | ||
| Name = {{PAGENAME}} | |||
| Image = Medulloblastoma with rosettes.jpg | |||
| Width = | |||
| Caption = Classic medulloblastoma [[H&E stain]]. | |||
| Synonyms = | |||
| Micro = | |||
| Subtypes = | |||
| LMDDx = small cell round blue tumous. | |||
| Stains = Reticulin +ve (in desmoplastic MB) | |||
| IHC = MAP2+ve | |||
| EM = | |||
| Molecular = | |||
| IF = | |||
| Gross = cerebellar. | |||
| Grossing = | |||
| Site = brain - usu. [[cerebellum]] | |||
| Assdx = | |||
| Syndromes = | |||
| Clinicalhx = | |||
| Signs = | |||
| Symptoms = | |||
| Prevalence = common - esp. in children | |||
| Bloodwork = | |||
| Rads = | |||
| Endoscopy = | |||
| Prognosis = subtype-dependent (WHO Grade IV) | |||
| Other = | |||
| ClinDDx = | |||
| Tx = | |||
}} | |||
'''Medulloblastoma''' is a [[malignant]] [[small round cell tumour]] that is found in the [[cerebellum]] or dorsal brainstem. It is the most common malignant CNS tumour in children. | |||
==General== | ==General== | ||
*Mostly paediatric population (mean age: 9 years). | *Mostly paediatric population (mean age: 9 years). | ||
*Tumors consists of histologically and molecular distinct subgroups. | |||
*All subgroups correspond to WHO grade IV. | |||
*May be seen as a component of [[nevoid basal cell carcinoma syndrome]] (NBCCS) AKA Gorlin syndrome, [[Li-Fraumeni syndrome]], Turcot syndrome, Rubinstain-Taybi syndrome and Nijmegen breakage syndrome. | *May be seen as a component of [[nevoid basal cell carcinoma syndrome]] (NBCCS) AKA Gorlin syndrome, [[Li-Fraumeni syndrome]], Turcot syndrome, Rubinstain-Taybi syndrome and Nijmegen breakage syndrome. | ||
**Show molecularly spatially homogeneous transcriptomes, allowing for accurate subgrouping of tumors from a single biopsy.<ref>{{Cite journal | last1 = Morrissy | first1 = AS. | last2 = Cavalli | first2 = FMG. | last3 = Remke | first3 = M. | last4 = Ramaswamy | first4 = V. | last5 = Shih | first5 = DJH. | last6 = Holgado | first6 = BL. | last7 = Farooq | first7 = H. | last8 = Donovan | first8 = LK. | last9 = Garzia | first9 = L. | title = Spatial heterogeneity in medulloblastoma. | journal = Nat Genet | volume = 49 | issue = 5 | pages = 780-788 | month = May | year = 2017 | doi = 10.1038/ng.3838 | PMID = 28394352 }}</ref> | **Show molecularly spatially homogeneous transcriptomes, allowing for accurate subgrouping of tumors from a single biopsy.<ref>{{Cite journal | last1 = Morrissy | first1 = AS. | last2 = Cavalli | first2 = FMG. | last3 = Remke | first3 = M. | last4 = Ramaswamy | first4 = V. | last5 = Shih | first5 = DJH. | last6 = Holgado | first6 = BL. | last7 = Farooq | first7 = H. | last8 = Donovan | first8 = LK. | last9 = Garzia | first9 = L. | title = Spatial heterogeneity in medulloblastoma. | journal = Nat Genet | volume = 49 | issue = 5 | pages = 780-788 | month = May | year = 2017 | doi = 10.1038/ng.3838 | PMID = 28394352 }}</ref> | ||
*Commonly spread via cerebrospinal fluid (CSF).<ref>{{Ref APBR|424 Q34}}</ref> | *Commonly spread via cerebrospinal fluid (CSF).<ref>{{Ref APBR|424 Q34}}</ref> | ||
**May be detected in [[CSF cytopathology]] specimens. | **May be detected in [[CSF cytopathology]] specimens. | ||
==Gross== | ==Gross== | ||
*Location: cerebellum - '''key feature''' or dorsal brainstem (lower rhombic lip). | *Location: cerebellum - '''key feature''' or dorsal brainstem (lower rhombic lip). | ||
** | **For morphologically identical supratentorial tumours see possible DDx. | ||
**Supratentorial and spinal metastases from initial | **Supratentorial and spinal metastases from initial medulloblastoma possible. | ||
==Microscopic== | ==Microscopic== | ||
Features:<ref>URL: [http://moon.ouhsc.edu/kfung/jty1/neurotest/Q93-Ans.htm http://moon.ouhsc.edu/kfung/jty1/neurotest/Q93-Ans.htm]. Accessed on: 26 October 2010.</ref> | Features:<ref>URL: [http://moon.ouhsc.edu/kfung/jty1/neurotest/Q93-Ans.htm http://moon.ouhsc.edu/kfung/jty1/neurotest/Q93-Ans.htm]. Accessed on: 26 October 2010.</ref> | ||
*[[Small round cell tumour]]. | *[[Small round cell tumour]]. | ||
*Mild to moderate nuclear pleomorphism. | |||
*High mitotic count. | |||
*Homer-Wright [[rosette]]s: | *Homer-Wright [[rosette]]s: | ||
**Rosette with a meshwork of fibers (neuropil) at the centre.<ref>{{cite journal |author=Wippold FJ, Perry A |title=Neuropathology for the neuroradiologist: rosettes and pseudorosettes |journal=AJNR Am J Neuroradiol |volume=27 |issue=3 |pages=488–92 |year=2006 |month=March |pmid=16551982 |doi= |url=}}</ref> | **Rosette with a meshwork of fibers (neuropil) at the centre.<ref>{{cite journal |author=Wippold FJ, Perry A |title=Neuropathology for the neuroradiologist: rosettes and pseudorosettes |journal=AJNR Am J Neuroradiol |volume=27 |issue=3 |pages=488–92 |year=2006 |month=March |pmid=16551982 |doi= |url=}}</ref> | ||
| Line 32: | Line 68: | ||
* Nestin +ve | * Nestin +ve | ||
* [[INI1]] retained (no loss) | * [[INI1]] retained (no loss) | ||
Genetic grouping with IHC:<ref>{{Cite journal | last1 = Pietsch | first1 = T. | last2 = Haberler | first2 = C. | title = Update on the integrated histopathological and genetic classification of medulloblastoma - a practical diagnostic guideline. | journal = Clin Neuropathol | volume = 35 | issue = 6 | pages = 344-352 | month = | year = | doi = 10.5414/NP300999 | PMID = 27781424 }}</ref> | |||
* WNT: beta-catein: nuclear +ve, YAP1+ve, OTX2+ve. | |||
* SHH: YAP1+ve, OTX2-ve, p75+ve. | |||
* Non-WNT/Non-SHH: * SHH: YAP1-ve, OTX2+ve, p75+ve. | |||
DDx: | DDx: | ||
*[[Small round cell tumours]]. | *[[Small round cell tumours]]. | ||
** [[AT/RT]] (INI1 loss) | ** [[AT/RT]]. (INI1 loss) | ||
** CNS-[[PNET]] | ** CNS-embryonal tumour (aka CNS-[[PNET]]). | ||
** ETMR (LIN28)-positive | ** ETMR (LIN28)-positive. | ||
** Ewing Sarcoma family members | ** Ewing Sarcoma family members. | ||
** Small cell glioblastoma | ** Small cell glioblastoma (Olig2 +ve) - esp. after RT and longer latency. <ref>{{Cite journal | last1 = Phi | first1 = JH. | last2 = Park | first2 = AK. | last3 = Lee | first3 = S. | last4 = Choi | first4 = SA. | last5 = Baek | first5 = IP. | last6 = Kim | first6 = P. | last7 = Kim | first7 = EH. | last8 = Park | first8 = HC. | last9 = Kim | first9 = BC. | title = Genomic analysis reveals secondary glioblastoma after radiotherapy in a subset of recurrent medulloblastomas. | journal = Acta Neuropathol | volume = | issue = | pages = | month = Apr | year = 2018 | doi = 10.1007/s00401-018-1845-8 | PMID = 29644394 }}</ref> | ||
** Anaplastic ependymoma. | |||
==Images== | ==Images== | ||
| Line 76: | Line 117: | ||
*#Desmoplastic/nodular medulloblastoma (DNMB). | *#Desmoplastic/nodular medulloblastoma (DNMB). | ||
*#Medulloblastoma with extensive nodularity (MBEN). | *#Medulloblastoma with extensive nodularity (MBEN). | ||
====Classic variant==== | |||
*Densely packed embryonal cells. | |||
*Mitosis / apoptosis present. | |||
*Nodules of neurocytic differentiation without internodular reticulin. | |||
*Rarely spongioblastic pattern (ribboning). | |||
====Anaplastic variant==== | ====Anaplastic variant==== | ||
| Line 93: | Line 140: | ||
===Genetically defined=== | ===Genetically defined=== | ||
*WNT (brainstem-related, usu. CTNNB1 mutations, less common: CSNK2B, AXIN2, APC, approx. 10%).<ref>{{Cite journal | last1 = Wefers | first1 = AK. | last2 = Warmuth-Metz | first2 = M. | last3 = Pöschl | first3 = J. | last4 = von Bueren | first4 = AO. | last5 = Monoranu | first5 = CM. | last6 = Seelos | first6 = K. | last7 = Peraud | first7 = A. | last8 = Tonn | first8 = JC. | last9 = Koch | first9 = A. | title = Subgroup-specific localization of human medulloblastoma based on pre-operative MRI. | journal = Acta Neuropathol | volume = 127 | issue = 6 | pages = 931-3 | month = | year = 2014 | doi = 10.1007/s00401-014-1271-5 | PMID = 24699697 }}</ref> | *WNT (brainstem-related, usu. CTNNB1 mutations, less common: CSNK2B, AXIN2, APC, approx. 10%).<ref>{{Cite journal | last1 = Wefers | first1 = AK. | last2 = Warmuth-Metz | first2 = M. | last3 = Pöschl | first3 = J. | last4 = von Bueren | first4 = AO. | last5 = Monoranu | first5 = CM. | last6 = Seelos | first6 = K. | last7 = Peraud | first7 = A. | last8 = Tonn | first8 = JC. | last9 = Koch | first9 = A. | title = Subgroup-specific localization of human medulloblastoma based on pre-operative MRI. | journal = Acta Neuropathol | volume = 127 | issue = 6 | pages = 931-3 | month = | year = 2014 | doi = 10.1007/s00401-014-1271-5 | PMID = 24699697 }}</ref> | ||
** Usu. classic MB in histology. | |||
** Caution: Some WNT-activated MB may show additional subclonal SHH-mutations.<ref>{{Cite journal | last1 = Iorgulescu | first1 = JB. | last2 = Van Ziffle | first2 = J. | last3 = Stevers | first3 = M. | last4 = Grenert | first4 = JP. | last5 = Bastian | first5 = BC. | last6 = Chavez | first6 = L. | last7 = Stichel | first7 = D. | last8 = Buchhalter | first8 = I. | last9 = Samuel | first9 = D. | title = Deep sequencing of WNT-activated medulloblastomas reveals secondary SHH pathway activation. | journal = Acta Neuropathol | volume = 135 | issue = 4 | pages = 635-638 | month = Apr | year = 2018 | doi = 10.1007/s00401-018-1819-x | PMID = 29435664 }}</ref> | |||
** Peak incidence: 7-14 years (15% adults). | |||
** Beta-catenin nuclear +ve. | |||
** Excellent prognosis. | |||
*SHH (PTCH1 & SUFU in infant, PTCH1, SMO, Gli2, MYCN in non-infant, approx 30%).<ref>{{Cite journal | last1 = Neumann | first1 = JE. | last2 = Swartling | first2 = FJ. | last3 = Schüller | first3 = U. | title = Medulloblastoma: experimental models and reality. | journal = Acta Neuropathol | volume = | issue = | pages = | month = Jul | year = 2017 | doi = 10.1007/s00401-017-1753-3 | PMID = 28725965 }}</ref> | *SHH (PTCH1 & SUFU in infant, PTCH1, SMO, Gli2, MYCN in non-infant, approx 30%).<ref>{{Cite journal | last1 = Neumann | first1 = JE. | last2 = Swartling | first2 = FJ. | last3 = Schüller | first3 = U. | title = Medulloblastoma: experimental models and reality. | journal = Acta Neuropathol | volume = | issue = | pages = | month = Jul | year = 2017 | doi = 10.1007/s00401-017-1753-3 | PMID = 28725965 }}</ref> | ||
** | **Origin in the cerebellar granule neuron cell precursors. | ||
*Group 3 (approx. 20%). | **Infantile and adult cases are biologically different, esp p53 mutant tumors are associated with poor outcome.<ref>{{Cite journal | last1 = Kool | first1 = M. | last2 = Jones | first2 = DT. | last3 = Jäger | first3 = N. | last4 = Northcott | first4 = PA. | last5 = Pugh | first5 = TJ. | last6 = Hovestadt | first6 = V. | last7 = Piro | first7 = RM. | last8 = Esparza | first8 = LA. | last9 = Markant | first9 = SL. | title = Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. | journal = Cancer Cell | volume = 25 | issue = 3 | pages = 393-405 | month = Mar | year = 2014 | doi = 10.1016/j.ccr.2014.02.004 | PMID = 24651015 }}</ref> | ||
*Group 4 (approx. 40%). | **Infantile, p53 wildtype: Usu. desmoplastic/nodular, 10q loss. | ||
** Group 3+4 | **SHH-p53 mutant: Usu. large cell/anaplastic, 17q loss. | ||
*Non WNT/Non SHH: | |||
**Group 3 (approx. 20%). | |||
***Classic and large cell/anaplastic, MYC amplification, isochromosome 17q. | |||
**Group 4 (approx. 40%). | |||
***Classic phenotype, MYCN amplification, isodicentric 17q. | |||
Note: Within Group 3+4 two or more of chromosome 7 gain, chromosome 8 loss, and chromosome 11 loss separates standard risk medulloblastoma samples into favorable and classifies the remaining i17q diploid cases as high-risk. | |||
==Prognosis== | ==Prognosis== | ||
Latest revision as of 15:20, 20 November 2019
| Medulloblastoma | |
|---|---|
| Diagnosis in short | |
 Classic medulloblastoma H&E stain. | |
| LM DDx | small cell round blue tumous. |
| Stains | Reticulin +ve (in desmoplastic MB) |
| IHC | MAP2+ve |
| Gross | cerebellar. |
| Site | brain - usu. cerebellum |
|
| |
| Prevalence | common - esp. in children |
| Prognosis | subtype-dependent (WHO Grade IV) |
Medulloblastoma is a malignant small round cell tumour that is found in the cerebellum or dorsal brainstem. It is the most common malignant CNS tumour in children.
General
- Mostly paediatric population (mean age: 9 years).
- Tumors consists of histologically and molecular distinct subgroups.
- All subgroups correspond to WHO grade IV.
- May be seen as a component of nevoid basal cell carcinoma syndrome (NBCCS) AKA Gorlin syndrome, Li-Fraumeni syndrome, Turcot syndrome, Rubinstain-Taybi syndrome and Nijmegen breakage syndrome.
- Show molecularly spatially homogeneous transcriptomes, allowing for accurate subgrouping of tumors from a single biopsy.[1]
- Commonly spread via cerebrospinal fluid (CSF).[2]
- May be detected in CSF cytopathology specimens.
Gross
- Location: cerebellum - key feature or dorsal brainstem (lower rhombic lip).
- For morphologically identical supratentorial tumours see possible DDx.
- Supratentorial and spinal metastases from initial medulloblastoma possible.
Microscopic
Features:[3]
- Small round cell tumour.
- Mild to moderate nuclear pleomorphism.
- High mitotic count.
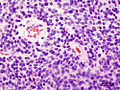
- Homer-Wright rosettes:
- Rosette with a meshwork of fibers (neuropil) at the centre.[4]
IHC
- MAP2 usu. +ve
- Synaptophysin +ve (weak to strong)
- NSE +ve/-ve
- NF +ve/-ve
- Chromogranin +ve/-ve
- GFAP +ve/-ve (mostly along blood vessels)
- Vimentin +ve
- Nestin +ve
- INI1 retained (no loss)
Genetic grouping with IHC:[5]
- WNT: beta-catein: nuclear +ve, YAP1+ve, OTX2+ve.
- SHH: YAP1+ve, OTX2-ve, p75+ve.
- Non-WNT/Non-SHH: * SHH: YAP1-ve, OTX2+ve, p75+ve.
DDx:
Images
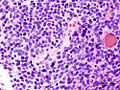
Medulloblastoma. (WC/KGH)
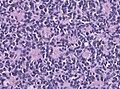
Medulloblastoma. (WC/KGH)
Homer-Wright rosettes in a medulloblastoma. (WC/jensflorian)
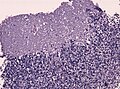
Cerebellar infiltrative growth. (WC/jensflorian)
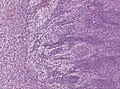
Demsomplastic nodular medullloblastoma. (WC/jensflorian)
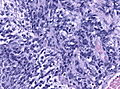
Anaplastic large cell medulloblastoma. (WC/jensflorian)
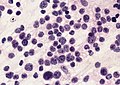
Medulloblastoma cells smear. (AFIP)
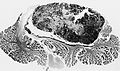
Medulloblastoma in the cerebellum. (AFIP)

Reticulin stain highlighting "pale islands" in a desmoplastic medulloblastoma (WC/jensflorian)
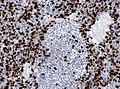
MIB-1 immunostaining in desmoplastic medulloblastoma (WC/jensflorian)
_in_adult.JPG)
_in_adult.JPG)







Case:
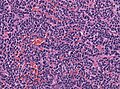
Classic Medulloblastoma of the SHH type. (WC/jensflorian)
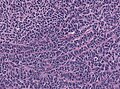
Epitheloid ribboning and nuclear moulding of tumor cells. (WC/jensflorian)
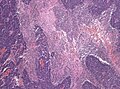
Areas of geographic necrosis. (WC/jensflorian)

Partial MAP2 immunoreactivity. (WC/jensflorian)



www:
- Medulloblastoma (ouhsc.edu).
- Medulloblastoma - several images (upmc.edu).
- Medulloblastoma with rhabdomyoblastic differentiation - several images (upmc.edu).
Subtypes
Histologically defined
- Classic medulloblastoma (~85% of all medulloblastomas).
- Variants of medulloblastoma (~15% of all medulloblastomas together):
- Anaplastic / Large cell variant.
- Desmoplastic/nodular medulloblastoma (DNMB).
- Medulloblastoma with extensive nodularity (MBEN).
Classic variant
- Densely packed embryonal cells.
- Mitosis / apoptosis present.
- Nodules of neurocytic differentiation without internodular reticulin.
- Rarely spongioblastic pattern (ribboning).
Anaplastic variant
Features:
- Larger cells.
- Severe anaplasia.
- Polygonal cells.
Desmoplastic/nodular variant
Features:
- Nodular, reticulin-free zones (pale islands).
- Mitotic activity is reduced.
- Densely packed internodal, reticulin-positive tumor areas.
- Absence of neuroblastic rosettes.
- Neuronal markers (NeuN, Synaptophysin) can be +ve.
Genetically defined
- WNT (brainstem-related, usu. CTNNB1 mutations, less common: CSNK2B, AXIN2, APC, approx. 10%).[7]
- Usu. classic MB in histology.
- Caution: Some WNT-activated MB may show additional subclonal SHH-mutations.[8]
- Peak incidence: 7-14 years (15% adults).
- Beta-catenin nuclear +ve.
- Excellent prognosis.
- SHH (PTCH1 & SUFU in infant, PTCH1, SMO, Gli2, MYCN in non-infant, approx 30%).[9]
- Origin in the cerebellar granule neuron cell precursors.
- Infantile and adult cases are biologically different, esp p53 mutant tumors are associated with poor outcome.[10]
- Infantile, p53 wildtype: Usu. desmoplastic/nodular, 10q loss.
- SHH-p53 mutant: Usu. large cell/anaplastic, 17q loss.
- Non WNT/Non SHH:
- Group 3 (approx. 20%).
- Classic and large cell/anaplastic, MYC amplification, isochromosome 17q.
- Group 4 (approx. 40%).
- Classic phenotype, MYCN amplification, isodicentric 17q.
- Group 3 (approx. 20%).
Note: Within Group 3+4 two or more of chromosome 7 gain, chromosome 8 loss, and chromosome 11 loss separates standard risk medulloblastoma samples into favorable and classifies the remaining i17q diploid cases as high-risk.
Prognosis
- Prognosis based on histology:[11][12][13] DNMB & MBEN > classic > anaplastic & large cell variant.
- Prognosis based on genetics:[14] WNT > SHH (without Tp53 mut) > Group 4 > Group 3 > SHH (with Tp53 mut).
See also
References
- ↑ Morrissy, AS.; Cavalli, FMG.; Remke, M.; Ramaswamy, V.; Shih, DJH.; Holgado, BL.; Farooq, H.; Donovan, LK. et al. (May 2017). "Spatial heterogeneity in medulloblastoma.". Nat Genet 49 (5): 780-788. doi:10.1038/ng.3838. PMID 28394352.
- ↑ Lefkowitch, Jay H. (2006). Anatomic Pathology Board Review (1st ed.). Saunders. pp. 424 Q34. ISBN 978-1416025887.
- ↑ URL: http://moon.ouhsc.edu/kfung/jty1/neurotest/Q93-Ans.htm. Accessed on: 26 October 2010.
- ↑ Wippold FJ, Perry A (March 2006). "Neuropathology for the neuroradiologist: rosettes and pseudorosettes". AJNR Am J Neuroradiol 27 (3): 488–92. PMID 16551982.
- ↑ Pietsch, T.; Haberler, C.. "Update on the integrated histopathological and genetic classification of medulloblastoma - a practical diagnostic guideline.". Clin Neuropathol 35 (6): 344-352. doi:10.5414/NP300999. PMID 27781424.
- ↑ Phi, JH.; Park, AK.; Lee, S.; Choi, SA.; Baek, IP.; Kim, P.; Kim, EH.; Park, HC. et al. (Apr 2018). "Genomic analysis reveals secondary glioblastoma after radiotherapy in a subset of recurrent medulloblastomas.". Acta Neuropathol. doi:10.1007/s00401-018-1845-8. PMID 29644394.
- ↑ Wefers, AK.; Warmuth-Metz, M.; Pöschl, J.; von Bueren, AO.; Monoranu, CM.; Seelos, K.; Peraud, A.; Tonn, JC. et al. (2014). "Subgroup-specific localization of human medulloblastoma based on pre-operative MRI.". Acta Neuropathol 127 (6): 931-3. doi:10.1007/s00401-014-1271-5. PMID 24699697.
- ↑ Iorgulescu, JB.; Van Ziffle, J.; Stevers, M.; Grenert, JP.; Bastian, BC.; Chavez, L.; Stichel, D.; Buchhalter, I. et al. (Apr 2018). "Deep sequencing of WNT-activated medulloblastomas reveals secondary SHH pathway activation.". Acta Neuropathol 135 (4): 635-638. doi:10.1007/s00401-018-1819-x. PMID 29435664.
- ↑ Neumann, JE.; Swartling, FJ.; Schüller, U. (Jul 2017). "Medulloblastoma: experimental models and reality.". Acta Neuropathol. doi:10.1007/s00401-017-1753-3. PMID 28725965.
- ↑ Kool, M.; Jones, DT.; Jäger, N.; Northcott, PA.; Pugh, TJ.; Hovestadt, V.; Piro, RM.; Esparza, LA. et al. (Mar 2014). "Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition.". Cancer Cell 25 (3): 393-405. doi:10.1016/j.ccr.2014.02.004. PMID 24651015.
- ↑ Gulino A, Arcella A, Giangaspero F (November 2008). "Pathological and molecular heterogeneity of medulloblastoma". Curr Opin Oncol 20 (6): 668–75. doi:10.1097/CCO.0b013e32831369f4. PMID 18841049.
- ↑ Rutkowski S, von Hoff K, Emser A, et al. (November 2010). "Survival and Prognostic Factors of Early Childhood Medulloblastoma: An International Meta-Analysis". J Clin Oncol 28 (33): 4961–4968. doi:10.1200/JCO.2010.30.2299. PMID 20940197.
- ↑ Rutkowski, S.; Bode, U.; Deinlein, F.; Ottensmeier, H.; Warmuth-Metz, M.; Soerensen, N.; Graf, N.; Emser, A. et al. (Mar 2005). "Treatment of early childhood medulloblastoma by postoperative chemotherapy alone.". N Engl J Med 352 (10): 978-86. doi:10.1056/NEJMoa042176. PMID 15758008.
- ↑ Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, SC.; Doz, F.; Kool, M.; Dufour, C. et al. (Jun 2016). "Risk stratification of childhood medulloblastoma in the molecular era: the current consensus.". Acta Neuropathol 131 (6): 821-31. doi:10.1007/s00401-016-1569-6. PMID 27040285.