Difference between revisions of "Gaucher disease"
Jump to navigation
Jump to search
| Line 28: | Line 28: | ||
| ClinDDx = | | ClinDDx = | ||
}} | }} | ||
'''Gaucher disease''' | '''Gaucher disease''' is the most common [[lysosomal storage diseases|lysosomal storage disease]].<ref name=pmid18466035>{{Cite journal | last1 = Chen | first1 = M. | last2 = Wang | first2 = J. | title = Gaucher disease: review of the literature. | journal = Arch Pathol Lab Med | volume = 132 | issue = 5 | pages = 851-3 | month = May | year = 2008 | doi = 10.1043/1543-2165(2008)132[851:GDROTL]2.0.CO;2 | PMID = 18466035 }}</ref> | ||
Despite being the most common in its grouping, it is still quite rare. Like most [[storage disorders]], it is inherited autosomal recessive; thus, it is seen more commonly in families where people are marry their cousins. | |||
==General== | ==General== | ||
Revision as of 02:21, 29 November 2013
| Gaucher disease | |
|---|---|
| Diagnosis in short | |
 Gaucher disease. H&E stain. | |
|
| |
| LM | crumpled tissue paper macrophages |
| Subtypes | type I, type II, type III |
| Site | bone, other |
|
| |
| Associated Dx | fracture of bone |
| Blood work | pancytopenia |
| Prognosis | dependent on subtype |
Gaucher disease is the most common lysosomal storage disease.[1]
Despite being the most common in its grouping, it is still quite rare. Like most storage disorders, it is inherited autosomal recessive; thus, it is seen more commonly in families where people are marry their cousins.
General
Pathology:
- Accumulation of glucocerebroside in monocytes/macrophages due to deficiency of glucocerebrosidase.[2]
- Defect in acid beta-glucosidase gene (GBA gene).[3][4][5]
Subtypes
- There are several types - all are autosomal recessive.[2]
Types:[6]
- Type I: 99% of cases; no CNS involvement - survive to adulthood.
- Type II: infantile onset - CNS degeneration + death at young age.
- Type III: mixed of type I & type II.
Clinical
- Pancytopenia - due to marrow replacement.
- Hepatosplenomegaly (type I).
Microscopic
- Mononuclear phagocytes with abundant eosinophilic cytoplasm with subtle irregular lines (~0.5 micrometers in width).
- Known as "crumpled tissue paper cells" / "crumpled tissue paper cytoplasm."[8]
Notes:
- Crumpled tissue paper: crumpled tissue paper - image (123rf.com).
- The textbook case may look crumpled... along with some mind altering drugs.
- The typical case is:
- Abundant macrophages with cytoplasm filled by very small (clear) vacuoles (~0.2-0.4 micrometres).
- The typical case is:
Images
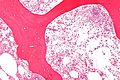
Gaucher disease - intermed. mag. (WC)

Gaucher disease - high mag. (WC)
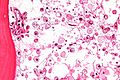
Gaucher disease - with fine vesicular cytoplasm - very high mag. (WC)


www:
- Gaucher disease - bone marrow aspirate (swmed.edu).
- Gaucher disease (webpathology.com).[7]
- Gaucher disease (neuropathologyweb.org).[9]
{kind=link}
Stains
- Material in "crumpled tissue paper cells": PAS +ve.[6]
See also
References
- ↑ Chen, M.; Wang, J. (May 2008). "Gaucher disease: review of the literature.". Arch Pathol Lab Med 132 (5): 851-3. doi:10.1043/1543-2165(2008)132[851:GDROTL]2.0.CO;2. PMID 18466035.
- ↑ 2.0 2.1 URL: http://emedicine.medscape.com/article/944157-overview. Accessed on: 3 December 2010.
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 230800
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 230900
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 231000
- ↑ 6.0 6.1 6.2 Mitchell, Richard; Kumar, Vinay; Fausto, Nelson; Abbas, Abul K.; Aster, Jon (2011). Pocket Companion to Robbins & Cotran Pathologic Basis of Disease (8th ed.). Elsevier Saunders. pp. 95. ISBN 978-1416054542.
- ↑ 7.0 7.1 URL: http://www.webpathology.com/image.asp?case=377&n=3. Accessed on: 30 November 2010.
- ↑ URL: http://pathcuric1.swmed.edu/pathdemo/gen1/gen130.htm. Accessed on: 28 May 2011.
- ↑ URL: http://www.neuropathologyweb.org/chapter10/chapter10bLSDs.html. Accessed on: 30 November 2010.